Devergie’s Lichen (Pityriasis Rubra Pilaris): Chronic Inflammatory Dermatosis
Overview
Devergie’s lichen, also known as pityriasis rubra pilaris (PRP), is a rare, chronic inflammatory skin disease of unknown origin. It is characterized by diffuse redness, scaling, and follicular hyperkeratosis (blockage and inflammation of hair follicles). Although the exact cause remains unclear, the disease is thought to involve an immunogenetic component. PRP affects individuals of all ages and both sexes equally.
Despite its rarity, PRP may cause significant physical and emotional discomfort due to its disfiguring nature and potential for systemic symptoms. The condition is non-infectious and is not transmitted between individuals, although familial cases with autosomal dominant inheritance have been documented.
Clinical Classification
There are six main clinical types of Devergie’s lichen, each with distinct epidemiological and morphological characteristics:
- Type I (Classic Adult Type): The most common variant, accounting for the majority of cases. It usually resolves spontaneously in 2–3 years and has the best prognosis.
- Type II (Atypical Adult Type): Represents approximately 5% of cases. It begins atypically, often without initial scalp involvement, and tends to be more chronic.
- Type III (Classic Juvenile Type): Accounts for around 10% of cases. It resembles Type I but occurs in children.
- Type IV (Circumscribed Juvenile Type): Represents about 25% of cases. Characterized by well-defined localized lesions, primarily on elbows and knees in children.
- Type V (Atypical Juvenile Type): A familial variant with autosomal dominant inheritance, manifesting early in life with mixed features of Types I and II.
- Type VI (HIV-Associated Type): Occurs in immunocompromised individuals, especially those with HIV. Presents with follicular-based plaques and often lacks classic palmoplantar involvement.
Symptoms and General Manifestations
The clinical presentation of Devergie’s lichen varies according to the subtype but typically includes:
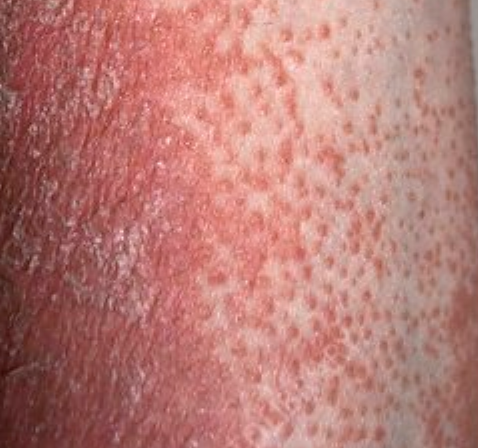
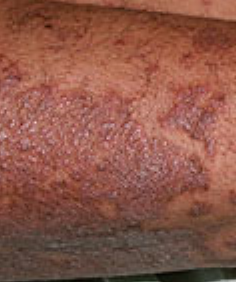
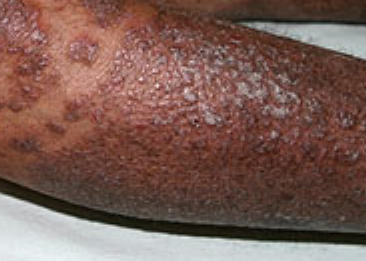
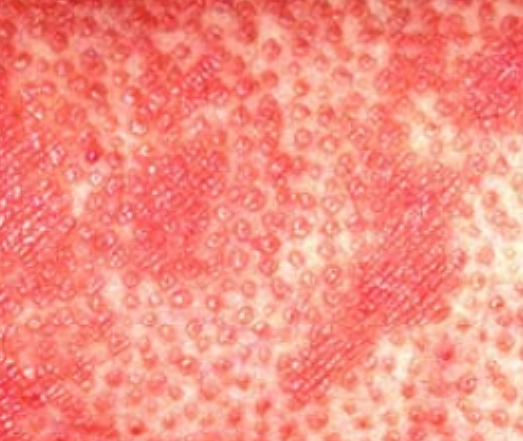
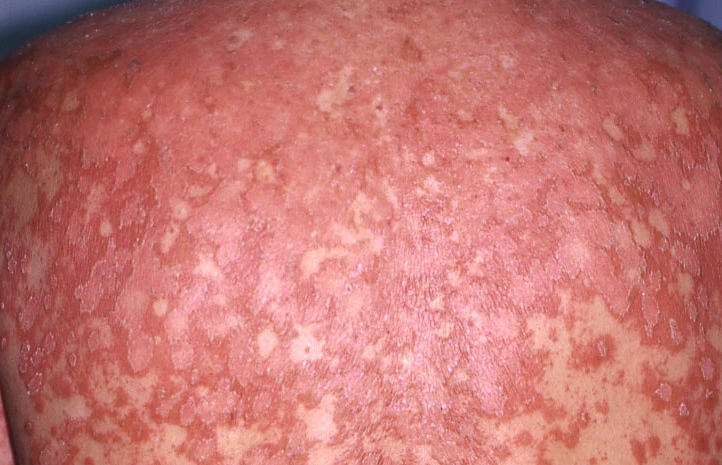
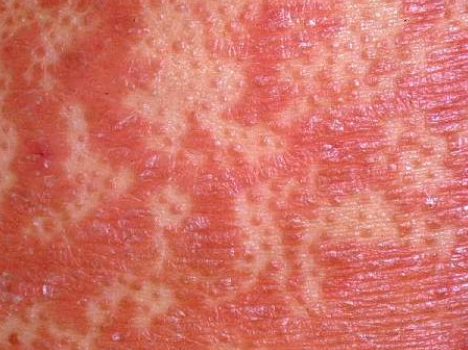
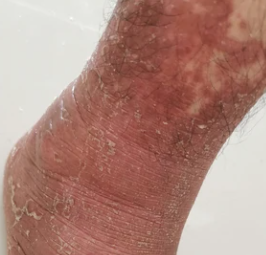
- Follicular papules: Small, rough papules centered around hair follicles, usually appearing in clusters;
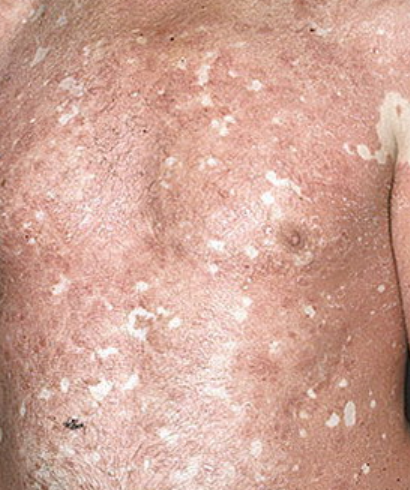
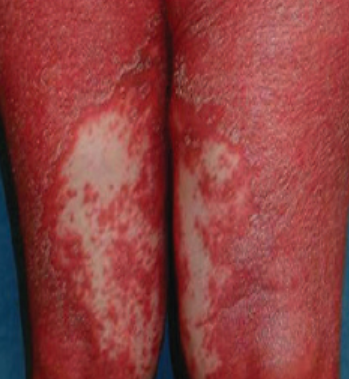
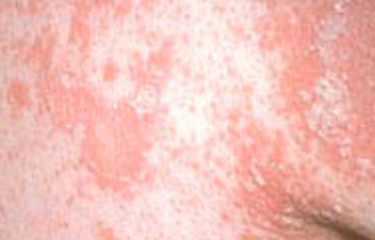
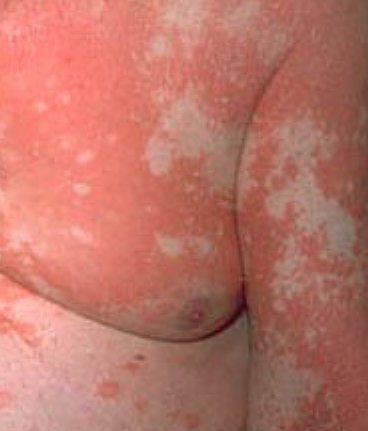
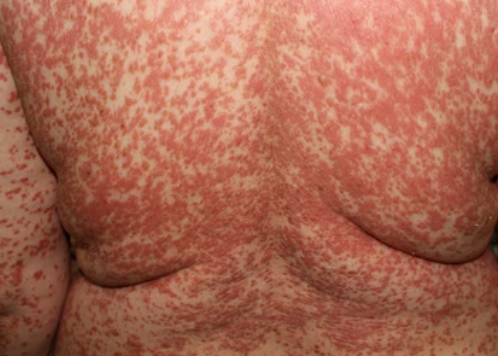
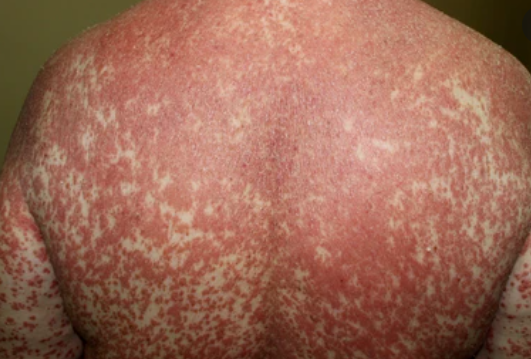
- Scaling and erythema: Diffuse red or orange-colored patches with dry, flaky skin;
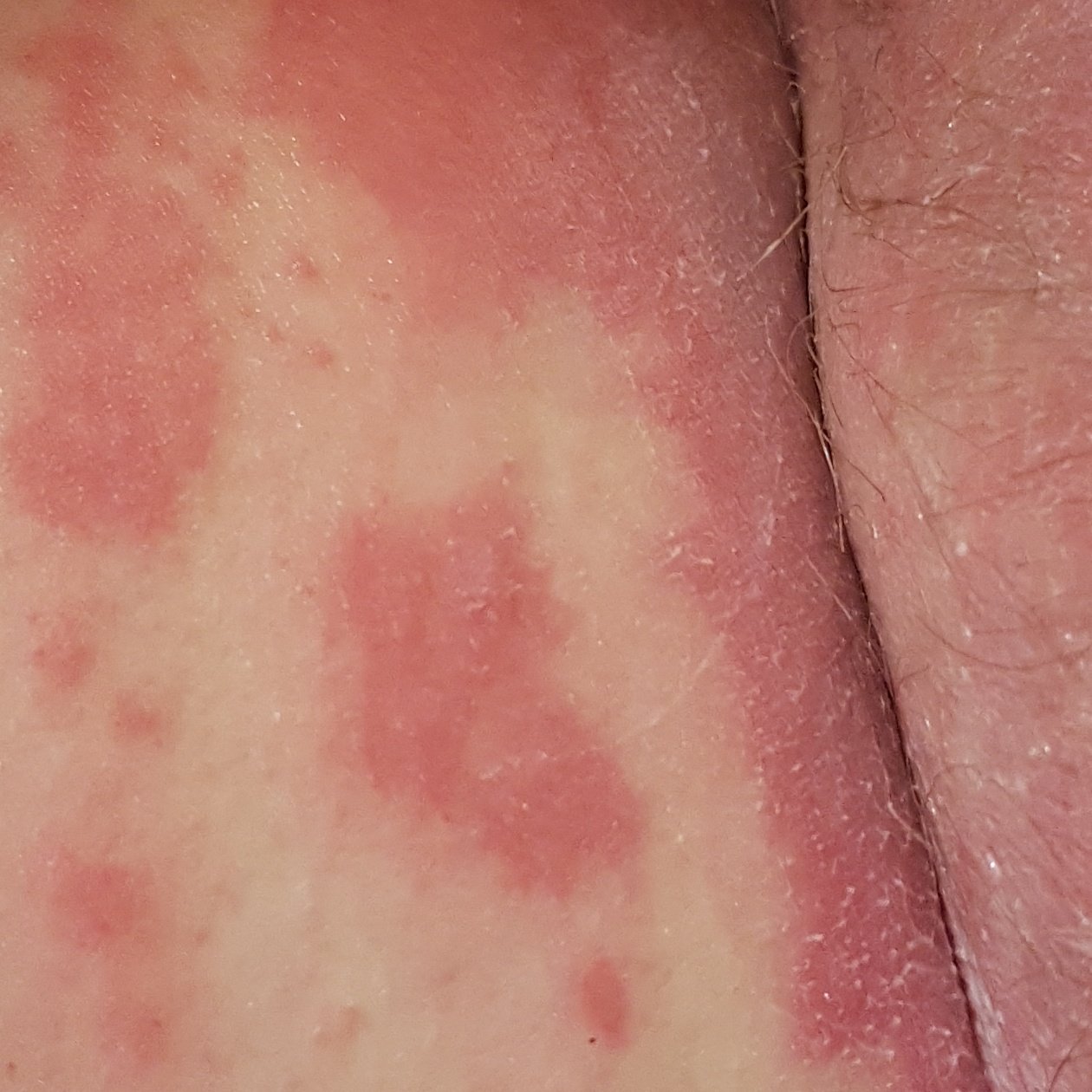
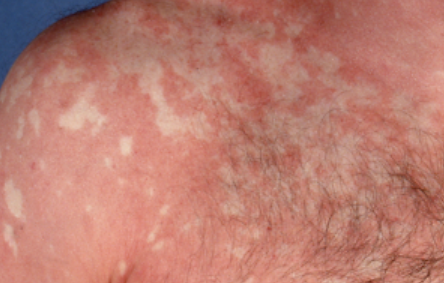
- Well-demarcated plaques: May coalesce and spread to large areas;
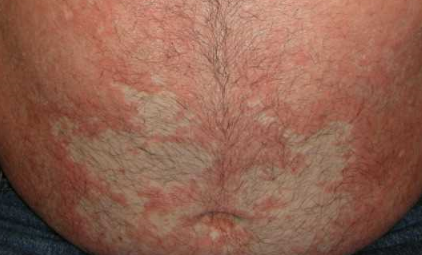
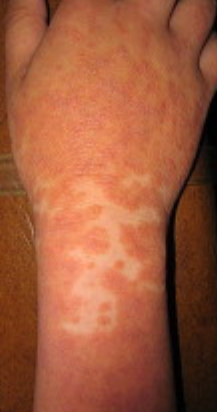
- “Islands of sparing”: Distinct areas of unaffected skin surrounded by inflammation;
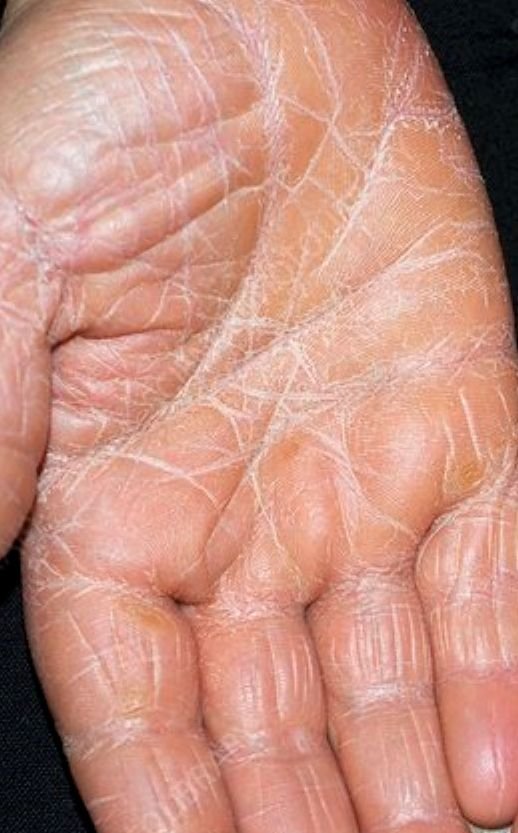
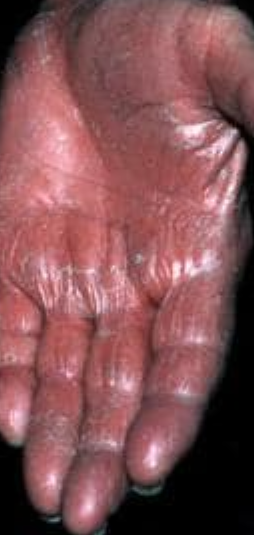
- Palmoplantar keratoderma: Thickening and orange discoloration of the skin on the palms and soles;
- Nail involvement: Thickened, discolored nails with longitudinal ridging or dystrophy in advanced stages;
- Scalp involvement: Scaly, erythematous plaques resembling psoriasis;
- Itching: Usually mild, but may worsen in progressive disease;
- Fatigue and fever: In severe or generalized forms, systemic symptoms may occur.
Detailed Clinical Subtypes of Devergie’s Lichen
Type I – Classic Adult Type
Begins abruptly, typically on the scalp, then spreads to the trunk and limbs. The lesions are dry, scaly, red-orange patches with follicular hyperkeratosis. Thickening of the palms and soles and nail changes may follow. This type may resolve spontaneously within 2–3 years in 80% of cases.
Type II – Atypical Adult Type
Represents around 5% of cases. Lacks the classic pattern; skin thickening on palms and soles may occur first, and hair loss can be prominent. It tends to be chronic and treatment-resistant.
Type III – Classic Juvenile Type
Accounts for ~10% of cases. Resembles Type I but appears in childhood. May show follicular papules and red-orange plaques, usually without systemic symptoms.
Type IV – Circumscribed Juvenile Type
Makes up ~25% of cases. Typically presents with well-defined localized lesions on elbows, knees, and palms/soles. Often seen in otherwise healthy children.
Type V – Atypical Juvenile Type (Familial)
Rare, with an autosomal dominant inheritance. Presents early in childhood with mixed signs of Types I and II. Often starts with follicular plugging and palmoplantar keratosis, with a slow progression.
Type VI – HIV-Associated
Seen in immunocompromised individuals. Involves follicular hyperkeratosis and red scaly plaques that may or may not affect the palms and soles. Frequently more refractory to treatment.
Diagnostics
The diagnosis is usually based on clinical features, but confirmation may require additional investigations in atypical or early-stage cases.
- Clinical examination: Identifies characteristic follicular papules, scaling, islands of sparing, and palmoplantar involvement.
- Skin biopsy: Confirms diagnosis. Histology typically reveals alternating orthokeratosis and parakeratosis in both vertical and horizontal directions (checkerboard pattern), follicular plugging, and perivascular lymphocytic infiltrate.
- KOH test and cultures: Performed to exclude fungal infections.
- Differential diagnosis: Psoriasis, ichthyosis, eczema, seborrheic dermatitis, and lymphoma must be ruled out.
Treatment of Devergie’s Lichen
There is no universally effective or standardized therapy for PRP. Treatment depends on disease severity and patient response. A combination of topical and systemic therapies is commonly used.
Topical Therapy:
- Topical corticosteroids: Reduce inflammation and erythema in localized lesions.
- Emollients and moisturizers: Essential to manage skin dryness and support barrier repair.
- Keratolytic agents: Salicylic acid or urea creams may be used to reduce scaling.
Systemic Therapy:
- Retinoids (acitretin, isotretinoin): Most commonly used systemic agents; reduce keratinization and inflammation.
- Methotrexate: An alternative immunosuppressive agent, useful in extensive or refractory disease.
- Systemic corticosteroids: Occasionally prescribed during acute flares, but not preferred long-term due to relapse upon withdrawal.
- Biologic therapies: In resistant cases, agents such as infliximab, adalimumab, ustekinumab, or secukinumab may be considered for targeting inflammatory cytokines.
Adjunctive Therapies:
- Antihistamines: Oral or topical agents for symptomatic relief of pruritus.
- Phototherapy (PUVA or narrowband UVB): May be effective in certain patients, but risk-benefit must be assessed.
Prognosis
The prognosis of Devergie’s lichen varies depending on the subtype. Type I often resolves spontaneously within 2–3 years, whereas Types II and VI tend to be more chronic and treatment-resistant. Juvenile forms may remit over time, but recurrence is possible. Long-term disease management often requires ongoing dermatologic supervision and supportive care.
Conclusion
Devergie’s lichen is a rare but impactful chronic skin disease with multiple clinical variants. It requires a high index of suspicion for diagnosis, especially in early stages or atypical cases. While there is no definitive cure, a combination of topical agents, systemic therapies, and supportive skin care allows for effective symptom control in most patients. Prompt recognition and early intervention can significantly improve patient outcomes and quality of life.