Lichen de Devergie (pityriasis rubra pilaris) : dermatose inflammatoire chronique
Aperçu
Le lichen de Devergie, également connu sous le nom de pityriasis rubra pilaris (PRP), est une maladie inflammatoire chronique rare de la peau d’origine inconnue. Elle se caractérise par une rougeur diffuse, une desquamation et une hyperkératose folliculaire (obstruction et inflammation des follicules pileux). Bien que la cause exacte reste incertaine, on pense que cette maladie implique une composante immunogénétique. Le PRP touche indifféremment les personnes de tout âge et des deux sexes.
Malgré sa rareté, le PRP peut entraîner une gêne physique et émotionnelle importante en raison de son caractère défigurant et de son potentiel de symptômes systémiques. Cette affection est non infectieuse et ne se transmet pas d’une personne à l’autre, bien que des cas familiaux avec transmission autosomique dominante aient été documentés.
Classification clinique
Il existe six types cliniques principaux de lichen de Devergie, chacun présentant des caractéristiques épidémiologiques et morphologiques distinctes :
- Type I (type adulte classique) : Variante la plus courante, représentant la majorité des cas. Elle disparaît généralement spontanément en 2 à 3 ans et présente le meilleur pronostic.
- Type II (type atypique chez l’adulte) : représente environ 5 % des cas. Il débute de manière atypique, souvent sans atteinte initiale du cuir chevelu, et a tendance à être plus chronique.
- Type III (type juvénile classique) : représente environ 10 % des cas. Il ressemble au type I, mais touche les enfants.
- Type IV (type juvénile circonscrit) : Représente environ 25 % des cas. Caractérisé par des lésions localisées bien définies, principalement sur les coudes et les genoux chez les enfants.
- Type V (type juvénile atypique) : Variante familiale à transmission autosomique dominante, se manifestant tôt dans la vie avec des caractéristiques mixtes des types I et II.
- Type VI (type associé au VIH) : survient chez les personnes immunodéprimées, en particulier celles atteintes du VIH. Se présente sous forme de plaques folliculaires et ne s’accompagne souvent pas d’une atteinte palmo-plantaire classique.
Symptômes et manifestations générales
La présentation clinique du lichen de Devergie varie selon le sous-type, mais comprend généralement :
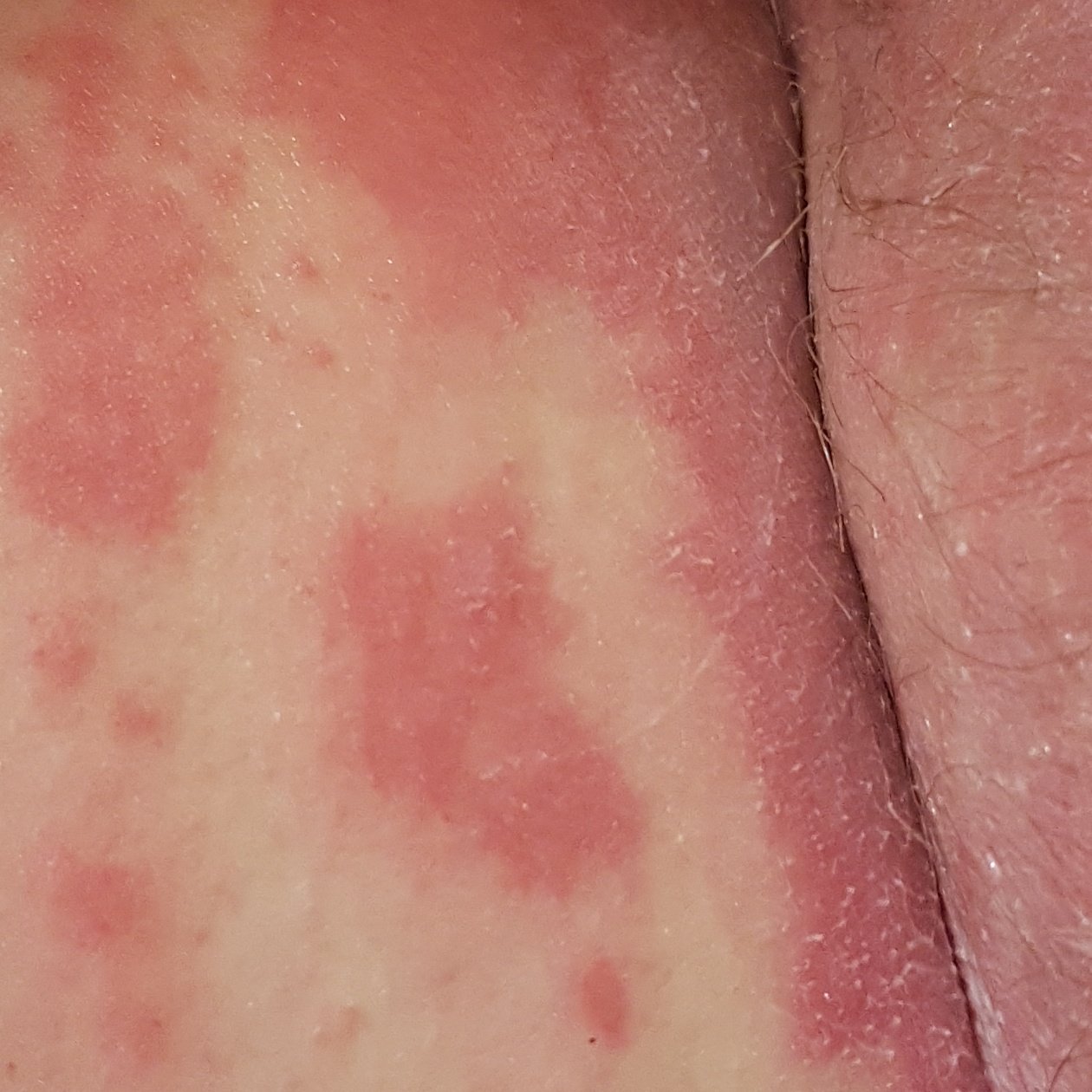
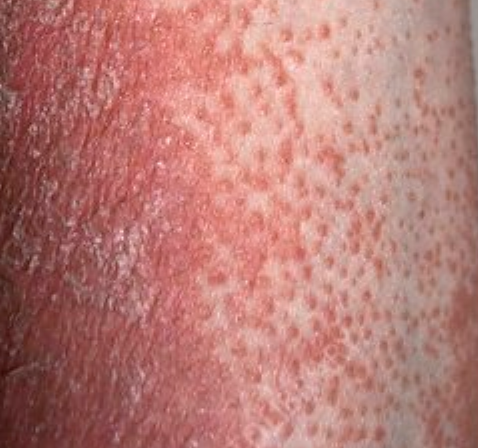
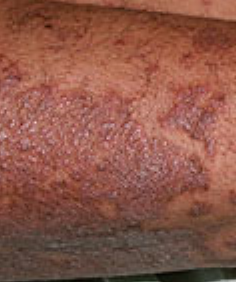
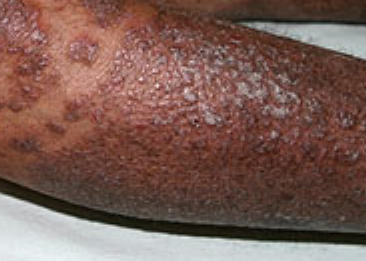
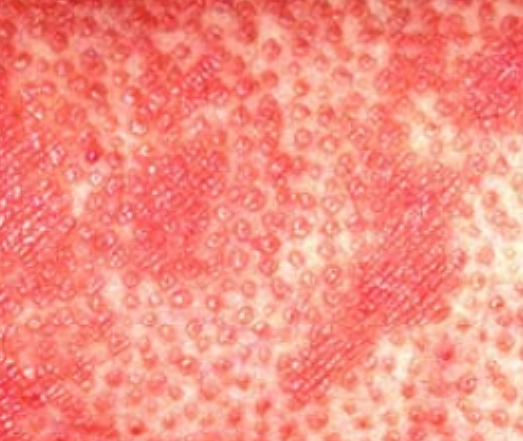
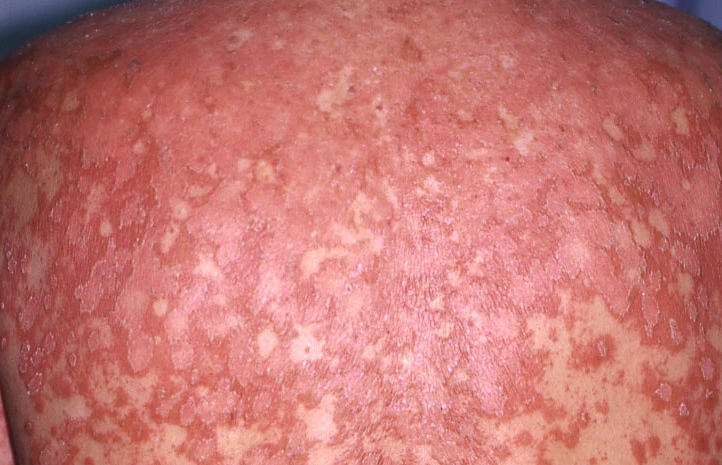
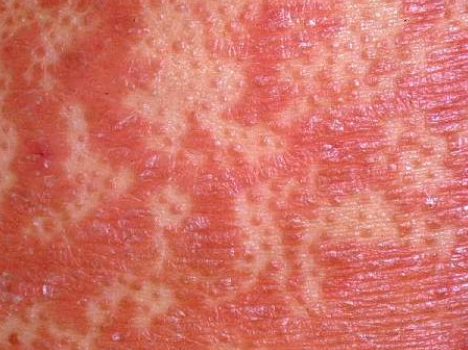
- Papules folliculaires : petites papules rugueuses centrées autour des follicules pileux, apparaissant généralement en grappes ;
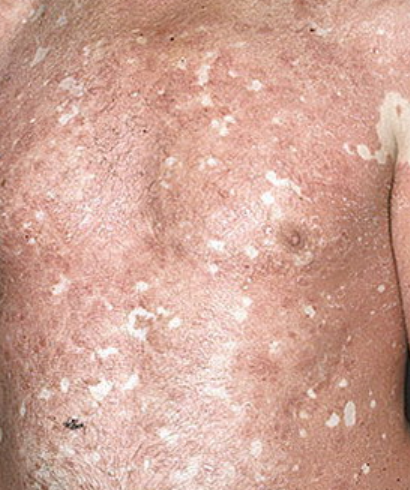
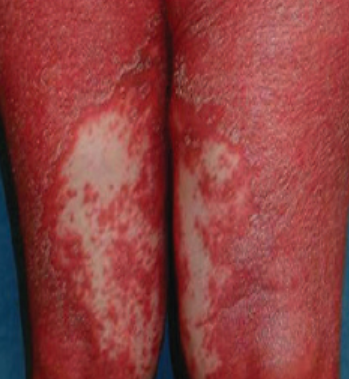
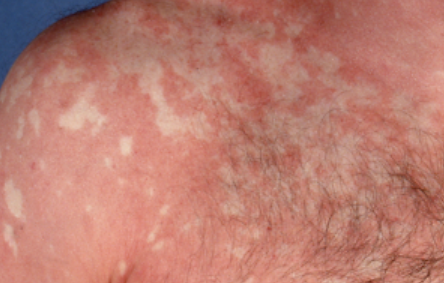
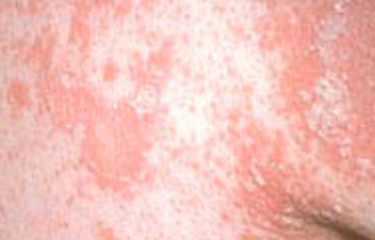
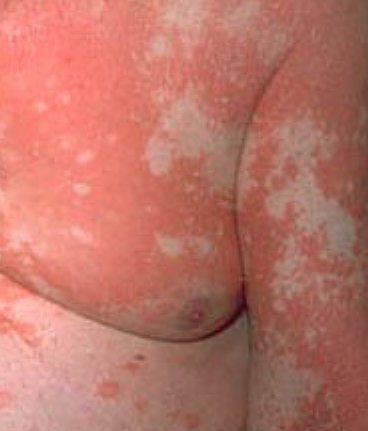
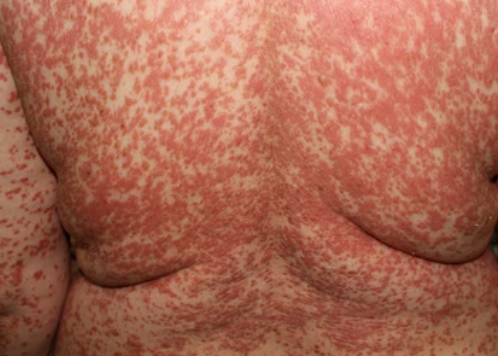
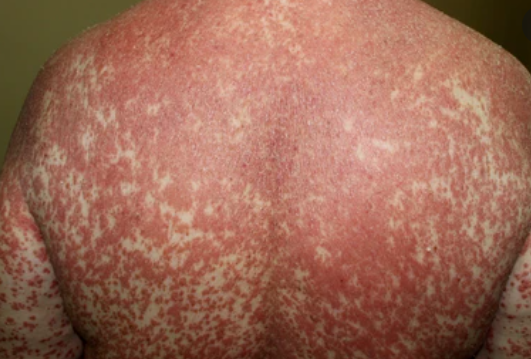
- Desquamation et érythème : plaques diffuses de couleur rouge ou orange avec une peau sèche et squameuse ;
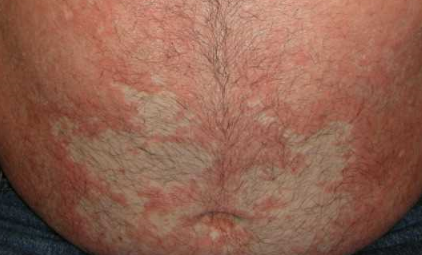
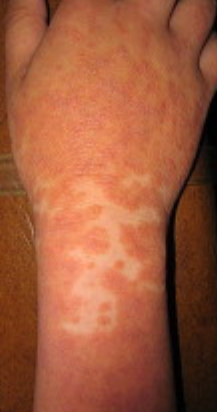
- Plaques bien délimitées : peuvent fusionner et s’étendre à de grandes zones ;
- « Îlots épargnés » : zones distinctes de peau non affectée entourées d’inflammation ;
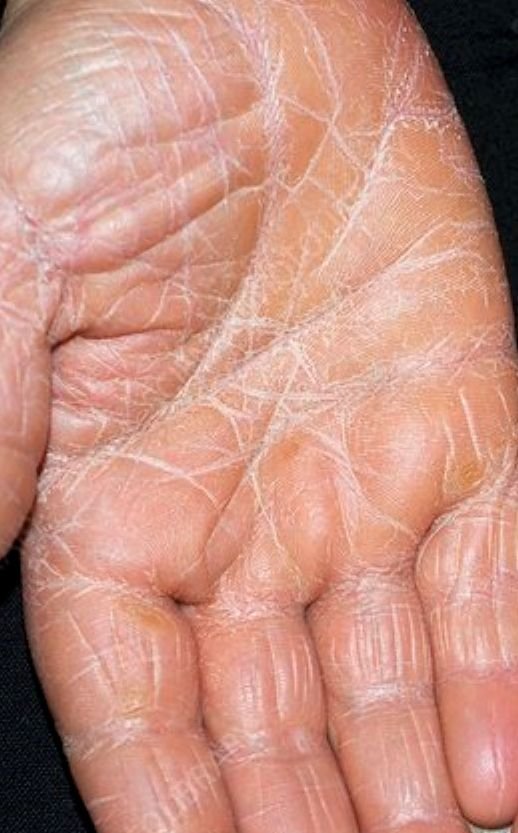
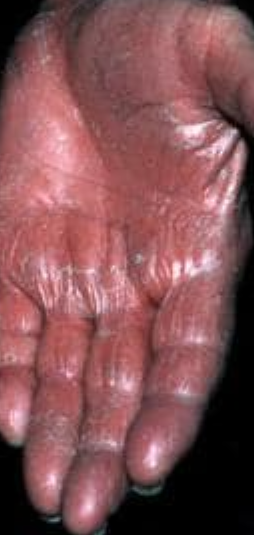
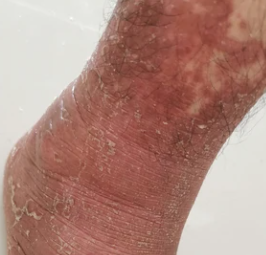
- Kératodermie palmo-plantaire : Épaississement et coloration orange de la peau des paumes et de la plante des pieds ;
- Atteinte des ongles : Ongles épaissis et décolorés, présentant des stries longitudinales ou une dystrophie à un stade avancé ;
- Atteinte du cuir chevelu : plaques squameuses et érythémateuses ressemblant à du psoriasis ;
- Démangeaisons : généralement légères, mais pouvant s’aggraver à mesure que la maladie progresse ;
- Fatigue et fièvre : dans les formes sévères ou généralisées, des symptômes systémiques peuvent apparaître.
Sous-types cliniques détaillés du lichen de Devergie
Type I – Type classique chez l’adulte
Apparaît brusquement, généralement sur le cuir chevelu, puis s’étend au tronc et aux membres. Les lésions sont sèches, squameuses, rouge-orange, avec une hyperkératose folliculaire. Un épaississement des paumes et de la plante des pieds et des modifications des ongles peuvent suivre. Ce type peut disparaître spontanément en 2 à 3 ans dans 80 % des cas.
Type II – Type adulte atypique
Représente environ 5 % des cas. Ne présente pas le schéma classique ; un épaississement de la peau au niveau des paumes et de la plante des pieds peut apparaître en premier, et une perte de cheveux peut être importante. Il a tendance à être chronique et résistant au traitement.
Type III – Type juvénile classique
Représente environ 10 % des cas. Ressemble au type I, mais apparaît pendant l’enfance. Peut présenter des papules folliculaires et des plaques rouge-orange, généralement sans symptômes systémiques.
Type IV – Type juvénile circonscrit
Représente environ 25 % des cas. Se manifeste généralement par des lésions localisées bien définies sur les coudes, les genoux et les paumes/plante des pieds. Souvent observée chez des enfants par ailleurs en bonne santé.
Type V – Type juvénile atypique (familial)
Rare, avec une hérédité autosomique dominante. Se manifeste tôt dans l’enfance avec des signes mixtes des types I et II. Commence souvent par un bouchon folliculaire et une kératose palmo-plantaire, avec une progression lente.
Type VI – Associé au VIH
Observé chez les personnes immunodéprimées. Se caractérise par une hyperkératose folliculaire et des plaques rouges squameuses qui peuvent ou non toucher les paumes et la plante des pieds. Souvent plus réfractaire au traitement.
Diagnostic
Le diagnostic repose généralement sur les signes cliniques, mais une confirmation peut nécessiter des examens complémentaires dans les cas atypiques ou à un stade précoce.
- Examen clinique : identifie les papules folliculaires caractéristiques, la desquamation, les îlots épargnés et l’atteinte palmo-plantaire.
- Biopsie cutanée : confirme le diagnostic. L’histologie révèle généralement une alternance d’orthokératose et de parakératose dans les directions verticale et horizontale (motif en damier), un bouchon folliculaire et une infiltration lymphocytaire périvasculaire.
- Test KOH et cultures : réalisés pour exclure les infections fongiques.
- Diagnostic différentiel : Le psoriasis, l’ichtyose, l’eczéma, la dermatite séborrhéique et le lymphome doivent être exclus.
Traitement du lichen de Devergie
Il n’existe pas de traitement universellement efficace ou standardisé pour le PRP. Le traitement dépend de la gravité de la maladie et de la réponse du patient. Une combinaison de traitements topiques et systémiques est couramment utilisée.
Traitement topique :
- Corticostéroïdes topiques : Réduisent l’inflammation et l’érythème dans les lésions localisées.
- Émollients et hydratants : essentiels pour traiter la sécheresse cutanée et favoriser la réparation de la barrière cutanée.
- Agents kératolytiques : des crèmes à base d’acide salicylique ou d’urée peuvent être utilisées pour réduire la desquamation.
Traitement systémique :
- Rétinoïdes (acitrétine, isotrétinoïne) : agents systémiques les plus couramment utilisés ; réduisent la kératinisation et l’inflammation.
- Méthotrexate : agent immunosuppresseur alternatif, utile dans les cas de maladie étendue ou réfractaire.
- Corticostéroïdes systémiques : Parfois prescrits lors de poussées aiguës, mais à éviter à long terme en raison du risque de rechute à l’arrêt du traitement.
- Thérapies biologiques : Dans les cas résistants, des agents tels que l’infliximab, l’adalimumab, l’ustekinumab ou le secukinumab peuvent être envisagés pour cibler les cytokines inflammatoires.
Traitements adjuvants :
- Antihistaminiques : Agents oraux ou topiques pour soulager les symptômes du prurit.
- Photothérapie (PUVA ou UVB à bande étroite) : Peut être efficace chez certains patients, mais le rapport bénéfice/risque doit être évalué.
Pronostic
Le pronostic du lichen de Devergie varie en fonction du sous-type. Le type I disparaît souvent spontanément en 2 à 3 ans, tandis que les types II et VI ont tendance à être plus chroniques et résistants au traitement. Les formes juvéniles peuvent régresser avec le temps, mais une récidive est possible. La prise en charge à long terme de la maladie nécessite souvent une surveillance dermatologique continue et des soins de soutien.
Conclusion
Le lichen de Devergie est une maladie cutanée chronique rare mais lourde qui présente de multiples variantes cliniques. Son diagnostic nécessite une grande vigilance, en particulier aux stades précoces ou dans les cas atypiques. Bien qu’il n’existe pas de traitement définitif, l’association d’agents topiques, de traitements systémiques et de soins cutanés de soutien permet de contrôler efficacement les symptômes chez la plupart des patients. Une reconnaissance rapide et une intervention précoce peuvent améliorer considérablement les résultats et la qualité de vie des patients.